

Highly parallel enhancer assays
As discussed on the Cis Regulatory Elements page, the in vivo reporter assays that have provided the traditional means of characterizing cell-type specific transcriptional enhancers are extremely labor-intensive and time-consuming. We have developed a new technology, termed ‘enhancer-FACS-Seq’ (eFS) (Gisselbrecht et al., Nature Methods, 2013), for highly parallel identification of active, tissue-specific transcriptional enhancers in the context of whole Drosophila embryos. As with traditional enhancer assays, each candidate CRM (cCRM) is cloned upstream of a reporter gene. Our key innovation is that we replace the use of microscopy to screen for tissue-specific enhancers with fluorescence activated cell sorting (FACS) of dissociated cells. This approach utilizes a two-marker system: in each embryo, one marker (here, the rat CD2 cell surface protein) is used to label cells of a specific tissue so that they can be sorted by FACS, and the other marker (here, green fluorescent protein [GFP]) is used as a reporter of CRM activity. Cells are sorted by tissue type and then by GFP fluorescence, and the cCRMs are recovered by PCR from double-positive sorted cells, and from total input cells. High-throughput sequencing of both populations then permits us to measure the relative abundance of each cCRM in input and sorted populations; we can then assess the enrichment or depletion of each cCRM in double-positive cells versus input as a measure of activity in the CD2-positive cell type being tested. By assessing enrichment in GFP-expressing CD2-negative as well as positive cells, and by crossing a common pool of reporter transformant male flies to females expressing CD2 in different cell types, we can assay specificity as well as activity.
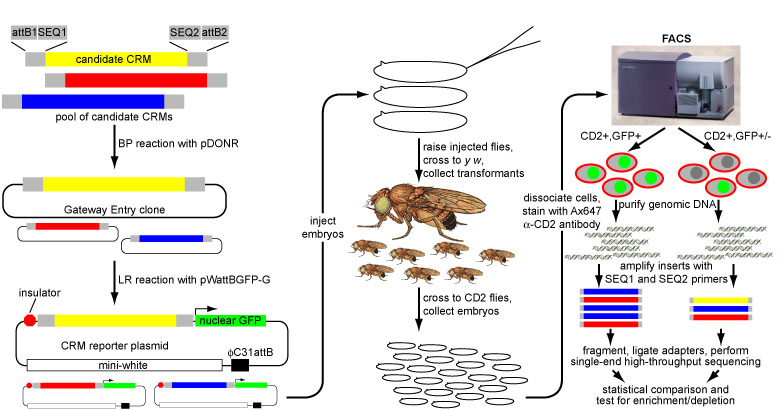
Overview of eFS experimental strategy.
In our initial report on this method (Gisselbrecht et al., Nature Methods, 2013), we generated a library of ~500 cCRMs drawn from a variety of genomic data sources (e.g,., TF-bound regions, coactivator-bound regions, DNaseI hypersensitive sites, and predictions from our PhylCRM algorithm) by PCR from genomic DNA, and screened them for activity in embryonic mesoderm and in specific mesodermal cell types. To validate our eFS results, we performed traditional reporter assays in Drosophila embryos for 68 cCRMs tested by eFS. The specificity of eFS was excellent among significantly enriched cCRMs, while sensitivity was good where the majority of the CD2-positive cells express GFP. As further validation, we found that the known enhancer-associated chromatin marks H3K27ac, H3K4me1, and Pol II are significantly enriched among the enhancers found to be active in mesoderm.
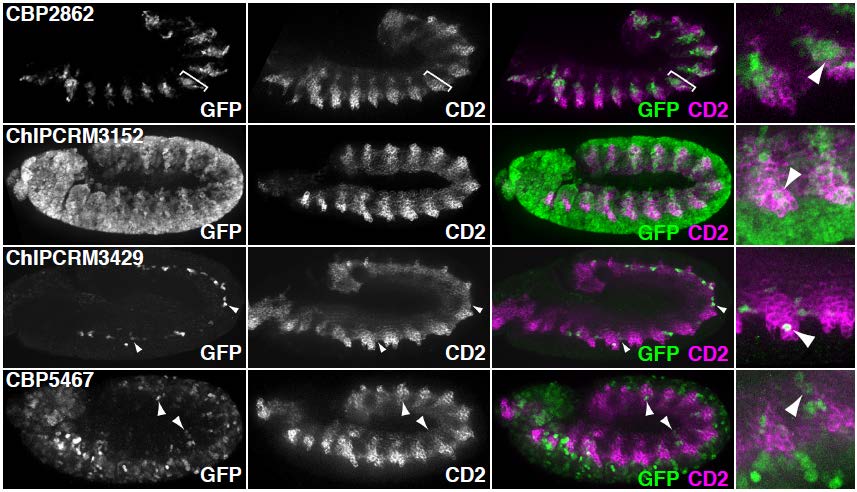
Validation of eFS. Mesodermal enhancers identified by eFS were tested for their ability to drive reporter gene expression (GFP) in cells expressing a mesodermal marker gene (CD2). Arrowheads highlight co-expressing cells.
Analysis of the eFS-annotated mesodermal enhancer sets enabled us to train a classifier to model whether cCRMs will be active or inactive in mesoderm or specifically in FCMs, based on the number and quality of matches to discriminatory TF binding motifs. These models performed better than ones based solely on previously known cis regulatory motifs for mesoderm and FCMs. We were also able to identify enriched TF binding motifs and motif combinations, potentially corresponding to novel mesodermal regulators and regulatory codes. For each of the 2 sets of eFS-positive cCRMs, we observed strong enrichment of the primary, known master regulator of that cell population: Twist for whole mesoderm, and Lameduck for FCMs. Motifs for other known mesodermal regulators were found in enriched combinations. We also saw strong enrichment of motifs for several sequence-specific DNA-binding proteins known to participate in recruitment of chromatin-modifying PcG and trxG proteins, suggesting a model in which regulatory competence of a noncoding region requires the confluence of binding sites for chromatin factors with those for tissue-specific TFs. Our lab is actively engaged in following up these putative novel regulators and cis regulatory motifs.
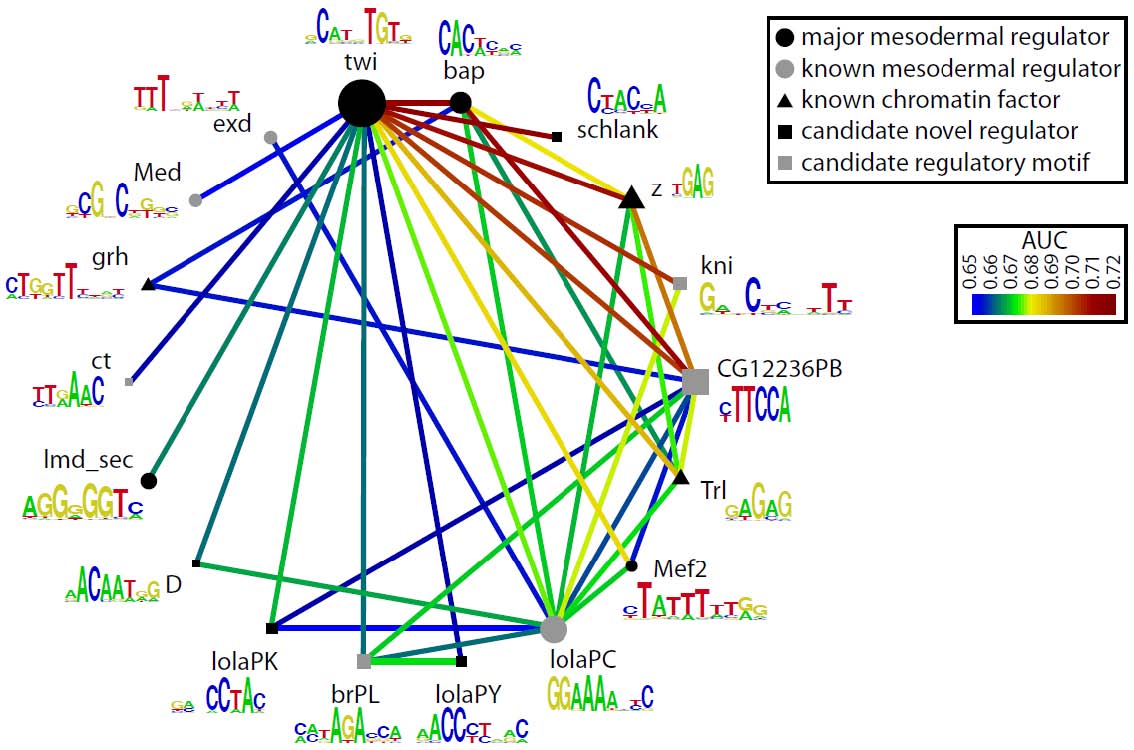
Candidate mesodermal regulatory codes from eFS data. Each edge represents a motif combination significantly enriched (q < 0.1, AUC > 0.65) in eFS-annotated mesodermal enhancers. Node diameter reflects enrichment of individual motifs.
By accelerating the annotation of the regulatory genome in Drosophila, we hope to generate the kind of high-volume regulatory interaction data that would allow us to explore the network properties of transcriptional regulation. In addition, we are actively engaged in trying to adapt and extend eFS to permit its use in other biological systems and for additional classes of cis regulatory features.