

Human Genetic Variation
Determining how genetic variation leads to variation in human traits is a central challenge for precision medicine. Mutations in either TFs or their DNA binding sites can perturb gene regulatory programs and lead to disease. Numerous TF mutations are known to cause developmental defects or adult-onset diseases, both rare and common. In addition, over 90% of variants found associated with complex traits in genome-wide association studies (GWAS), lie within the noncoding portions of the human genome.
Coding Variation
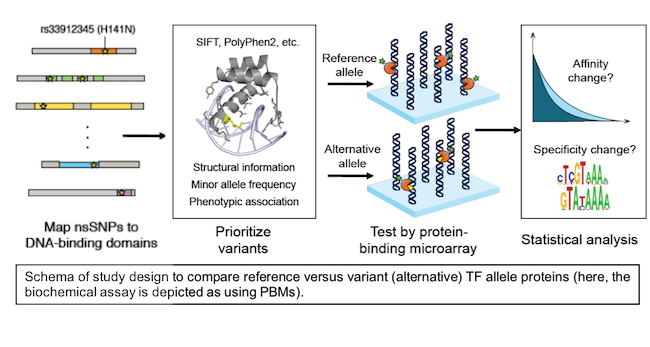 Disease mutations and naturally occurring coding variation in TFs, especially those within DBDs, have the potential to disrupt TF DNA binding activity. However, there is insufficient understanding of the determinants of TF-DNA binding activity to allow for accurate prediction of the effects of such coding mutations on DNA binding activity. Exponentially increasing amounts of genetic variation data from genotyping and exome- and whole-genome sequencing present a significant interpretation challenge, and genome interpretation tools have not focused on predicting the effects of coding variation in TFs. This is an important problem, since mutations or coding variation that damage TF DNA binding activity have the potential to disrupt the regulation of thousands of genes in the human genome.
Disease mutations and naturally occurring coding variation in TFs, especially those within DBDs, have the potential to disrupt TF DNA binding activity. However, there is insufficient understanding of the determinants of TF-DNA binding activity to allow for accurate prediction of the effects of such coding mutations on DNA binding activity. Exponentially increasing amounts of genetic variation data from genotyping and exome- and whole-genome sequencing present a significant interpretation challenge, and genome interpretation tools have not focused on predicting the effects of coding variation in TFs. This is an important problem, since mutations or coding variation that damage TF DNA binding activity have the potential to disrupt the regulation of thousands of genes in the human genome.
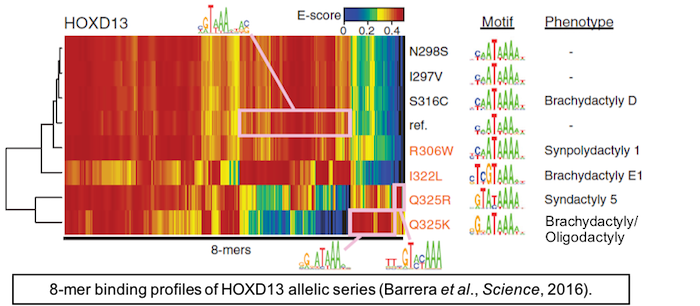 In Barrera et al., Science (2016), we analyzed genotype data for over 64,000 individuals and identified over 58,000 unique mutations that perturb DBDs.
We also analyzed known Mendelian disease mutations in TFs annotated as causing a wide range of diseases including developmental disorders.
We developed a computational approach to prioritize variants likely to damage TF DNA binding activity, which we then used to identify thousands of variants predicted to alter DNA binding activity.
Mendelian disease mutants and coding variants found in the genotyped populations both exhibited a spectrum of effects on DNA binding affinity and/or specificity, as revealed by protein-binding microarray (PBM) assays.
Altered occupancies of genomic target sites in ChIP-Seq data and dysregulation of the associated target genes from RNA-Seq data were consistent with mutants’ altered PBM profiles. Intriguingly, while individual DBD alleles predicted to damage DNA binding activity are rare, in aggregate they are prevalent: our results suggest that most unrelated individuals harbor a unique repertoire of TF alleles with a distinct trans -regulatory collective of DNA-binding activities.
In Barrera et al., Science (2016), we analyzed genotype data for over 64,000 individuals and identified over 58,000 unique mutations that perturb DBDs.
We also analyzed known Mendelian disease mutations in TFs annotated as causing a wide range of diseases including developmental disorders.
We developed a computational approach to prioritize variants likely to damage TF DNA binding activity, which we then used to identify thousands of variants predicted to alter DNA binding activity.
Mendelian disease mutants and coding variants found in the genotyped populations both exhibited a spectrum of effects on DNA binding affinity and/or specificity, as revealed by protein-binding microarray (PBM) assays.
Altered occupancies of genomic target sites in ChIP-Seq data and dysregulation of the associated target genes from RNA-Seq data were consistent with mutants’ altered PBM profiles. Intriguingly, while individual DBD alleles predicted to damage DNA binding activity are rare, in aggregate they are prevalent: our results suggest that most unrelated individuals harbor a unique repertoire of TF alleles with a distinct trans -regulatory collective of DNA-binding activities.
We are analyzing genome sequence data from various cohorts of pediatric patients with structural birth defects, with the goal of identifying genetic variants that damage the ability of transcription factors to recognize their DNA target sequences. We anticipate our results will help to identify pathogenic variants contributing to structural birth defects, reveal mechanisms by which such variants may dysregulate gene expression leading to these disorders and lead to refined genomic diagnostics.
Our results are revealing how different alleles of the same TF (i.e., different coding variants) exert different effects on DNA binding, and are opening new areas of investigation into studies of how TF coding variation impacts transcriptional gene regulation and phenotypes.
Noncoding Variation
 We are developing strategies that employ data from DNA binding assays, as well as other functional genomic assays, to interpret the effects of noncoding variants for their potential to perturb transcriptional regulation.
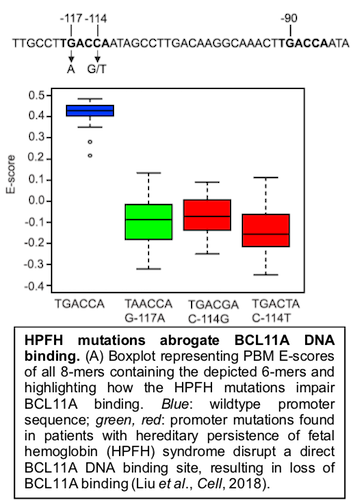
In our collaborative Liu et al., Cell (2018) paper with Dr. Stuart Orkin’s lab, we showed how universal PBM data can reveal the mechanisms responsible for the effects of clinical noncoding regulatory variants found in individuals with hereditary persistence of fetal hemoglobin (HPFH) syndrome.
Our PBM analysis of the TF BCL11A, which acts to repress γ-globin gene expression, revealed that mutations found in the promoters of the human γ-globin genes in HPFH individuals abrogate binding by BCL11A.
We are developing strategies that employ data from DNA binding assays, as well as other functional genomic assays, to interpret the effects of noncoding variants for their potential to perturb transcriptional regulation.
In our collaborative Liu et al., Cell (2018) paper with Dr. Stuart Orkin’s lab, we showed how universal PBM data can reveal the mechanisms responsible for the effects of clinical noncoding regulatory variants found in individuals with hereditary persistence of fetal hemoglobin (HPFH) syndrome.
Our PBM analysis of the TF BCL11A, which acts to repress γ-globin gene expression, revealed that mutations found in the promoters of the human γ-globin genes in HPFH individuals abrogate binding by BCL11A.